This clinical brief was developed by Cardiometabolic Health Congress (CMHC) and supported by an educational grant from Alnylam Pharmaceuticals, Inc. Click Here to claim up to 0.50 CE/CME credits by 9/19/2023.
Introduction
Transthyretin (TTR) cardiac amyloidosis (ATTR-CM) is a restrictive cardiomyopathy that is caused by misfolding of transthyretin protein due to either aging (also called wild type, or ATTRwt) or a genetic mutation (called hereditary or mutant, ATTRm). [1] TTR misfolding leads to its accumulation in the myocardium, thickening of both ventricles, and subsequent restrictive cardiomyopathy and systolic dysfunction. [1]
Transthyretin amyloid cardiomyopathy is a progressive disease, and although prognosis is more favorable compared to other amyloid cardiomyopathies, ATTRm is associated with a poor prognosis if left untreated. [1] Historically, ATTR-CM was thought to be a rare cause of heart failure, but recent data have shown that it is actually more common, especially in people of African descent. [2] Additionally, ATTR-CM disproportionally affects Blacks and African Americans; particularly, ATTRm due to a Val122Ile mutation of the TTR gene affects up to 4% of African-Americans and is associated with a 16% 4-year survival rate, a median survival rate of 26 months, and high rates of cardiovascular hospitalizations. [2-4]
Due to low awareness and difficulty in distinguishing this condition from hypertensive, hypertrophic heart disease, patients with ATTR-CM are often misdiagnosed and diagnostic delays are common. [1] Additionally, these delays are exacerbated in Blacks and African Americans despite the increased disease burden, and these populations continue to be underrepresented in clinical trials. [2] Up until recently, there were no FDA-approved therapies for ATTR-CM, but the recent approval of tafamidis and the other agents that are in development, have the potential to address the historically limited treatment options for this disease. [1,5]
Pathophysiology of ATTR-CM
ATTR-CM is characterized by the buildup of misfolded transthyretin (TTR) protein that deposits in the cardiac tissue as amyloid.1 TTR is a plasma transport protein synthesized primarily in the liver, and its function is to transport thyroid hormone and vitamin A. [6] The wild-type entity is thought to be caused by the aging process, which affects the stability of the TTR protein, causing it to misfold and aggregate, although the exact mechanisms are unknown. [6] The wild-type ATTR predominantly affects the heart, however extracardiac soft tissue depositions may occur. [6]
ATTRwt is more common in males and prevalence increases with age, however, a recent study showed a higher prevalence of this subset in females than previously published. [7] Hereditary single-point genetic mutations in the TTR gene can alter protein structure and promote amyloid formation, and in addition to cardiac involvement, peripheral neuropathy can be common.1 Hereditary ATTR-CM is transmitted in an autosomal dominant pattern. Although there are more than 100 single-point mutations in the TTR gene that can lead to several TTR-related amyloid syndromes, the most common mutations are Val30Met, Thr60Ala, and Val122Ile.8 The first two are more prevalent in the European populations and Japan, while Val122Ile is the most prevalent cause of ATTRm in North America, with up to 4% of African-Americans carrying this mutation in the TTR gene. [1]
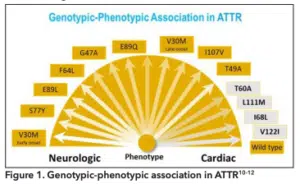
The Val122Ile form of ATTRm is associated with greater cardiac consequences compared to the other subsets of ATTRm, and as such, performing a detailed family history in individuals of African descent is important to the timely identification of this condition. [1,9] There is a clear genotypic-phenotypic correlation in ATTR, with the V122l mutation as well as wtATTR more closely associated with the cardiac vs. neurologic manifestations of the condition (Figure 1).[10-12]
Clinical Cues, Screening, and Diagnosis
Patients with ATTR cardiac amyloidosis initially may not show many symptoms, and the disease does not exhibit the early aggressive course that characterizes another predominant form of amyloidosis, immunoglobulin light-chain (AL). [1] ATTR-CM may present with a wide range of clinical cues, both cardiac and non-cardiac, as outlined in a recent scientific statement from the American Heart Association (Figure 2). [9,13]

However, as disease progresses, patients exhibit signs indicative of heart failure, and with progression of disease, congestive heart failure with reduced ejection fraction becomes apparent. [1] In addition, atrial fibrillation, angina pectoris, and myocardial infarction may occur in ATTR patients. [1] Several other symptoms cited above may be red flags for cardiac amyloidosis, such as intolerance to beta-blockers or angiotensin-converting enzyme (ACE) inhibitors, sudden normal or low blood pressure in patients with a history of hypertension, history of bilateral carpal tunnel syndrome, and mild increases in troponin levels. [8]
Other important characteristics or scenarios that may raise suspicion for ATTR particularly include a family history of ATTRm amyloidosis, African-American ancestry (≥60 years of age) with heart failure with preserved ejection fraction (HFpEF) and no hypertension, history of spinal stenosis, and a diagnosis of hypertrophic cardiomyopathy in an elderly patient, as well as some other clues mentioned above. [8,9,13] Traditionally, ATTR-CM is believed to be associated with heart failure with preserved ejection fraction (HFpEF), however, in a recent study, 45% of patients diagnosed with ATTR-CM presented with a left ventricular ejection fraction (LVEF) of ≤50%, with racial disparities in the LVEF at presentation in both variant and WT ATTR. [14] The study suggested that clinicians should extend their clinical suspicion for diagnosis of ATTR-CM beyond HFpEF presentation, especially in African American patients. [14]
Additionally, black patients with the Val122Ile mutation tend to be older, with a lower LVEF, and greater atrial dilation on echocardiography. [15] A more recent study suggested that compared to Caucasian patients, African American patients with hereditary ATTR-CM have a lower burden of atrial fibrillation.16 In individuals with a high index of suspicion for ATTR-CM based on the clues mentioned above, further diagnostic testing is recommended. Until recently, an invasive diagnostic approach by obtaining a biopsy was viewed as the gold standard, but advances in non-invasive diagnostics have expanded these options. A non-invasive diagnostic approach using bone scintigraphy and laboratory tests to exclude a diagnosis of LA are recommended, however, if laboratory tests suggest the presence of AL amyloidosis, bone scintigraphy should not be used to make a diagnosis of ATTR-CM and a biopsy is recommended in this setting. [13]
Novel nuclear imaging techniques, such as bone scintigraphy with technetium (Tc)-labeled bisphosphonates can be used to track TTR cardiac amyloid deposits, although the exact mechanisms for this remains unknown. [13] 99mTc-PYP (pyrophosphate) and 99mTc-DPD (3,3-diphosphono-1,2-propa- nodiacarboxylicacid) scintigraphy, and 99mTc-HMDP (hydroxymethylene diphosphonate) have shown efficacy in identifying ATTR cardiac amyloidosis, with a sensitivity of >99% and specificity of 86% across studies. [13]
Laboratory tests that measure serum free light chain (sFLC) are necessary to exclude a diagnosis of LA. [8] Despite advances in non-invasive techniques, obtaining a tissue biopsy to confirm amyloid deposition by Congo red staining, and more specific techniques (immunohistochemistry, mass spectrometry) can be performed to analyze the type of fibril present and differentiate between AL and ATTR. [1] If TTR fibrils are confirmed, genetic testing can be performed to differentiate between ATTRwt and ATTRm. [6,9]
Current and emerging treatment options for ATTR-CM
Current and emerging treatments for ATTR-CM work by either stabilizing, silencing, or disruption of TTR:
- TTR Stabilization. One approach to treat ATTR is to create small-molecules that stabilize the TTR protein to inhibit the rate-determining step of amyloidogenesis. [8] Diflunisal is a generic non-steroidal anti-inflammatory drug (NSAID) that binds to the thyroxine binding sites of TTR, preventing dissociation and fibril formation. [6] It has been shown to reduce progression to neuropathy and preserve quality of life in familial amyloid polyneuropathy (FAP), but it may not be tolerated by many ATTR patients. [17,18]
Tafamidis is a novel small-molecule TTR stabilizer, which also interacts with the thyroxine binding pocket and increases TTR stability. [6] Tafamidis has been shown to stabilize TTR, improve quality of life and echocardiographic parameters in ATTRm patients in a phase 2 study. [19] The landmark phase 3 trial with this agent, ATTR-ACT, evaluated its efficacy and safety in ATTR-CM (mutant or wildtype) patients; with tafamidis treatment significantly reducing all-cause mortality and frequency of cardiovascular-related hospitalization compared to placebo. [20]
Recent post-hoc analysis from this trial have shown that treatment with tafamidis significantly reduced the decline in 6-minute walk test (6MWT) and improve the patients’ quality of life, assessed by the Kansas City Cardiomyopathy Questionnaire overall score (KCCQ-OS); these effects were observed in both wild-type and mutant patients. [21,22] In the ATTR-ACT trial, approximately 14% of population enrolled were Black. [23] Based on these results, tafamidis was approved by the FDA in May 2019 for the treatment of adults with either wildtype or mutated ATTR-CM, making it the first and only therapy to date for this indication in the US. [5]
Anther TTR stabilizer, acoramidis (AG10), which has been shown to stabilize wild-type and mutant TTR in pre-clinical studies, [24] is currently being evaluated in a phase 2 trial in both wild-type and mutant ATTR. [25] A phase 2 trial evaluating the efficacy and safety of AG10 in ATTR-CM (mutant or wild type) patients with symptomatic, chronic heart failure showed that AG10 treatment effectively and safely restored serum TTR to the normal range in all subjects in a dose-dependent manner. [26] As a result, a phase 3 trial with this agent in ATTR-CM is underway with the primary outcomes being change from baseline 6MWT and total number of deaths due to all-cause and frequency of cardiovascular-related hospitalizations, with results expected by 2023. [27]
- TTR Suppression/Silencing. Another approach is suppression of TTR expression by silencing translation of TTR protein with small interfering RNA (siRNA) or degradation of TTR m-RNA by anti-sense oligonucleotides. Patisiran, an siRNA agent that specifically blocks liver TTR synthesis, was recently shown in the phase 3 APOLLO trial to improve neuropathy, as well as quality of life, walking, and nutritional status in patients with ATTRm. [28] A phase 3 study to evaluate the long-term efficacy and safety of patisiran is currently ongoing. [29] Based on the APOLLO trial, patisiran was approved by the FDA in 2018 for the treatment of polyneuropathy caused by ATTRm, which did not include a cardiovascular indication. A sub-analysis of the cardiac subpopulation of the APOLLO trial (56% of the total study population) showed that patisiran decreased mean left ventricular wall thickness, global longitudinal strain, NT-proBNP, as well as reductions in cardiac hospitalizations and all-cause mortality; suggesting that this agent may have also potential for the treatment of ATTR-CM.30
A phase 3 trial to assess the efficacy of patisiran in ATTR-CM are ongoing, including the APOLLO-B trial.31 Recently, it was announced that patisiran showed positive topline results from the APOLLO-B trial, but the results were not available at the time of writing of this document. Another investigational siRNA agent, vutrisiran, is undergoing phase 3 trials for hATTR polyneuropathy (for which it recently received a fast-track designation by the FDA) and hATTR-CM as part of the HELIOS A and B trials.32 The hATTR-CM phase III trial will evaluate the efficacy and safety of vutrisiran on all-cause mortality and frequency of cardiovascular hospitalizations.32 The ongoing phase 3 HELIOS-B study will evaluate the efficacy and safety of vutrisiran in patients with ATTR-CM (both wtATTR-CM and hATTR-CM), with results expected by 2024-2025.33 Inotersen, an anti-sense oligonucleotide, has been shown to reduce TTR production in a phase I trial, and a phase III trial in patients with hereditary ATTR-CM was recently concluded. In this trial, inotersen was deemed safe in patients with ATTR-CM.34 In addition, inotersen was FDA approved in 2018 for the treatment of polyneuropathy caused by ATTRm, and a study evaluating its efficacy in ATTR-CM (both mutant and wild-type) is ongoing.35 Preliminary results from this study have shown that inotersen has the potential to improve cardiac parameters (stabilization of LVEF, longitudinal strain and BNP, and improvement in 6MWT).35 Eplontersen (formerly AKCEA-TTR-LRX), an antisense oligonucleotide in development for patients with hereditary and wild-type ATTR, is currently in clinical development, including a specific phase 3 study, CARDIO-TTRansform, in patients with ATTR-CM.36 Additionally, this agent is being evaluated in another ongoing phase 3 study, NEURO-TTRansform, which is looking at patients with polyneuropathy.37
- TTR Disruption. Disruption of TTR is another strategy for ATTR-CM treatment, however, this approach is in its early stages of development compared to others. Pre-clinical studies have suggested that doxycycline, tauro-ursodeoxycholic acid (TUDCA) and natural polyphenols can disrupt amyloid fibrils and reduce aggregation, however, further studies are needed.6 GSK2315698, a serum amyloid depleter, was being evaluated in a phase II trial in patients with cardiac amyloidosis including ATTR, which was terminated due to safety concerns.38 Another agent, PRX004, the first anti-amyloid immunotherapy for ATTR-CM, is also in development.39
Conclusion
ATTR cardiac amyloidosis is a complex disease that remains challenging to diagnose and treat, and disproportionally affects Black and African American patients. Low awareness about the disease, and limited therapeutic options are significant barriers to optimal care, however, significant advances have been made in the development of novel targeted-therapeutics that may improve patient outcomes. Additionally, advances have been made to identify ATTR-CM noninvasively, and an accurate and early diagnosis is key to enabling appropriate care. In addition, medications to reduce symptoms and reduce impact are recommended due to the progressive nature of the disease. Given these advances and the continued research in ATTR-CM, it is important that clinicians are aware of the impacts and disparities that exist in ATTR-CM diagnosis and treatment to help optimize management and improve outcomes.
References
- Mankad, Anit K., and Keyur B. Shah. “Transthyretin cardiac amyloidosis.” Current cardiology reports 19.10 (2017): 97.
- Spencer-Bonilla, Gabriela, et al. “Racial and Ethnic Disparities in Transthyretin Cardiac Amyloidosis.” Current Cardiovascular Risk Reports 15.6 (2021): 1-8.
- Arruda-Olson, Adelaide M., et al. “Genotype, echocardiography, and survival in familial transthyretin amyloidosis.” Amyloid 20.4 (2013): 263- 268.
- Ruberg, Frederick L., et al. “Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS).” American heart journal 164.2 (2012): 222-228.
- Rubin, Jonah, and Mathew S. Maurer. “Cardiac Amyloidosis: Overlooked, Underappreciated, and Treatable.” Annual Review of Medicine 71 (2020): 203-219.
- Siddiqi, Omar K., and Frederick L. Ruberg. “Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment.” Trends in cardiovascular medicine (2017).
- González-López, Esther, et al. “Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths.” European heart journal 38.24 (2017): 1895-1904.
- Donnelly, J. P., and M. Hanna. “Cardiac amyloidosis: An update on diagnosis and treatment.” Cleveland Clinic journal of medicine 84.12 Suppl 3 (2017): 12-26.
- Kittleson, Michelle M., et al. “Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association.” Circulation 142.1 (2020): e7-e22.
- Connors, Lawreen H., et al. “Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis.” American heart journal 158.4 (2009): 607-614.
- Dungu, Jason N. “Cardiac amyloid–an update.” European Cardiology Review 10.2 (2015): 113.
- Rapezzi, Claudio, et al. “Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective.” European heart journal 34.7 (2013): 520-528.
- Witteles, Ronald M., et al. “Screening for transthyretin amyloid cardiomyopathy in everyday practice.” JACC: Heart Failure 7.8 (2019): 709-716.
- Martyn, Trejeeve M., et al. “A Substantial Proportion of Patients With Transthyretin Cardiac Amyloid (ATTR) Present With Impaired Left Ventricular Ejection Fraction: Implications for Diagnosis and Race Disparities.” Circulation 144.Suppl_1 (2021): A13409-A13409.
- Shah, Keyur B., et al. “Transthyretin cardiac amyloidosis in black Americans.” Circulation: Heart Failure 9.6 (2016): e002558.
- Mitrani, Lindsey, et al. “AFRICAN AMERICANS PRESENT WITH LESS ATRIAL FIBRILLATION IN TRANSTHYRETIN CARDIAC AMYLOIDOSIS: A RETROSPECTIVE REVIEW.” Journal of the American College of Cardiology 75.11_Supplement_1 (2020): 892-892.
- Berk, John L., et al. “Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial.” Jama 310.24 (2013): 2658-2667.
- Castaño, Adam, et al. “Diflunisal for ATTR cardiac amyloidosis.” Congestive heart failure 18.6 (2012): 315-319.
- Merlini, Giampaolo, et al. “Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis.” Journal of cardiovascular translational research 6.6 (2013): 1011-1020.
- Maurer, Mathew S., et al. “Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy.” New England Journal of Medicine 379.11 (2018): 1007-1016.
- Grogan, M., et al. “Efficacy of Tafamidis in Patients with Hereditary or Wild-Type Transthyretin Amyloid Cardiomyopathy: Further Results from the ATTR-ACT Trial.” The Journal of Heart and Lung Transplantation 38.4 (2019): S204.
- Rapezzi, Claudio, et al. “Efficacy of tafamidis in patients with hereditary and wild-type transthyretin amyloid cardiomyopathy: further analyses from ATTR-ACT.” Heart Failure 9.2 (2021): 115-123.
- Singh, Bishnu Mohan, et al. “A Systematic Review of Tafamidis in Patients With Transthyretin Amyloid Cardiomyopathy.” Cureus 13.9 (2021).
- Penchala, Sravan, et al. “Evaluation of AG10 as a Potential Therapy for Transthyretin Cardiac Amyloidosis.” (2015).
- ClinicalTrials.gov, NCT03536767, available at https://clinicaltrials. gov/ct2/show/NCT03536767, accessed August 9, 2022.
- Judge, Daniel P., et al. “Transthyretin Stabilization by AG10 in Symptomatic Transthyretin Amyloid Cardiomyopathy.” Journal of the American College of Cardiology (2019).
- Gillmore, Julian D., et al. “ATTRibute-CM: A Randomized, Double-Blind, Placebo-Controlled, Multi-Center, Global Phase 3 Study of AG10 in Patients With Transthyretin Amyloid Cardiomyopathy (ATTR-CM).” Circulation 140.Suppl_1 (2019): A14214-A14214.
- Adams, David, et al. “Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis.” New England Journal of Medicine 379.1 (2018): 11-21.
- ClinicalTrials.gov, NCT02510261. Available at https://clinicaltrials.gov/ct2/show/NCT02510261, accessed August 9, 2022.
- Solomon, Scott D., et al. “Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients With Hereditary Transthyretin-Mediated Amyloidosis: Analysis of the APOLLO Study.” Circulation 139.4 (2019): 431-443.
- ClinicalTrials.gov, NCT03997383. Available at https://www.clinicaltrials.gov/ct2/show/NCT03997383, accessed August 9, 2022.
- Shilling, Rebecca, et al. “STUDY DESIGN AND RATIONALE OF HELIOS-B: A PHASE 3 STUDY TO EVALUATE THE CLINICAL EFFICACY AND SAFETY OF VUTRISIRAN IN PATIENTS WITH ATTR AMYLOIDOSIS WITH CARDIOMYOPATHY.” Journal of the American College of Cardiology 75.11 Supplement 1 (2020): 3579.
- ClinicalTrials.gov, NCT04153149. Available at https://clinicaltrials.gov/ct2/show/NCT04153149, accessed August 9, 2022.
- Dasgupta, Noel R., et al. “SAFETY OF INOTERSEN TREATMENT IN PATIENTS WITH TRANSTHYRETIN AMYLOID CARDIOMYOPATHY.” Journal of the American College of Cardiology 73.9 Supplement 1 (2019): 909.
- Dasgupta, Noel Rakhi, and Merrill Benson. “IMPROVED SURVIVAL OF PATIENTS WITH TRANSTHYRETIN AMYLOID CARDIOMYOPATHY WITH INOTERSEN (TTR SPECIFIC ANTISENSE OLIGONUCLEOTIDE).” Journal of the American College of Cardiology 73.9 Supplement 1 (2019): 811.
- ClinicalTrials.gov, NCT04136171. Available at https://clinicaltrials.gov/ct2/show/NCT04136171, accessed August 9, 2022.
- ClinicalTrials.gov, NCT04136184. Available at https://clinicaltrials.gov/ct2/show/NCT04136184, accessed August 9, 2022.
- ClinicalTrials.gov, NCT03044353. Available at https://clinicaltrials.gov/ct2/show/NCT03044353, accessed August 9, 2022.
- Yadav, Jankhna D., et al. “Transthyretin Amyloid Cardiomyopathy— Current and Future Therapies.” Annals of Pharmacotherapy (2021): 10600280211000351.